Which mutations are amenable to exon 51 skipping?1-3
For exon skipping to work, exon 51 must be present in the patient's DMD gene.
The common DMD deletions that are theoretically amenable to exon 51 skipping include:
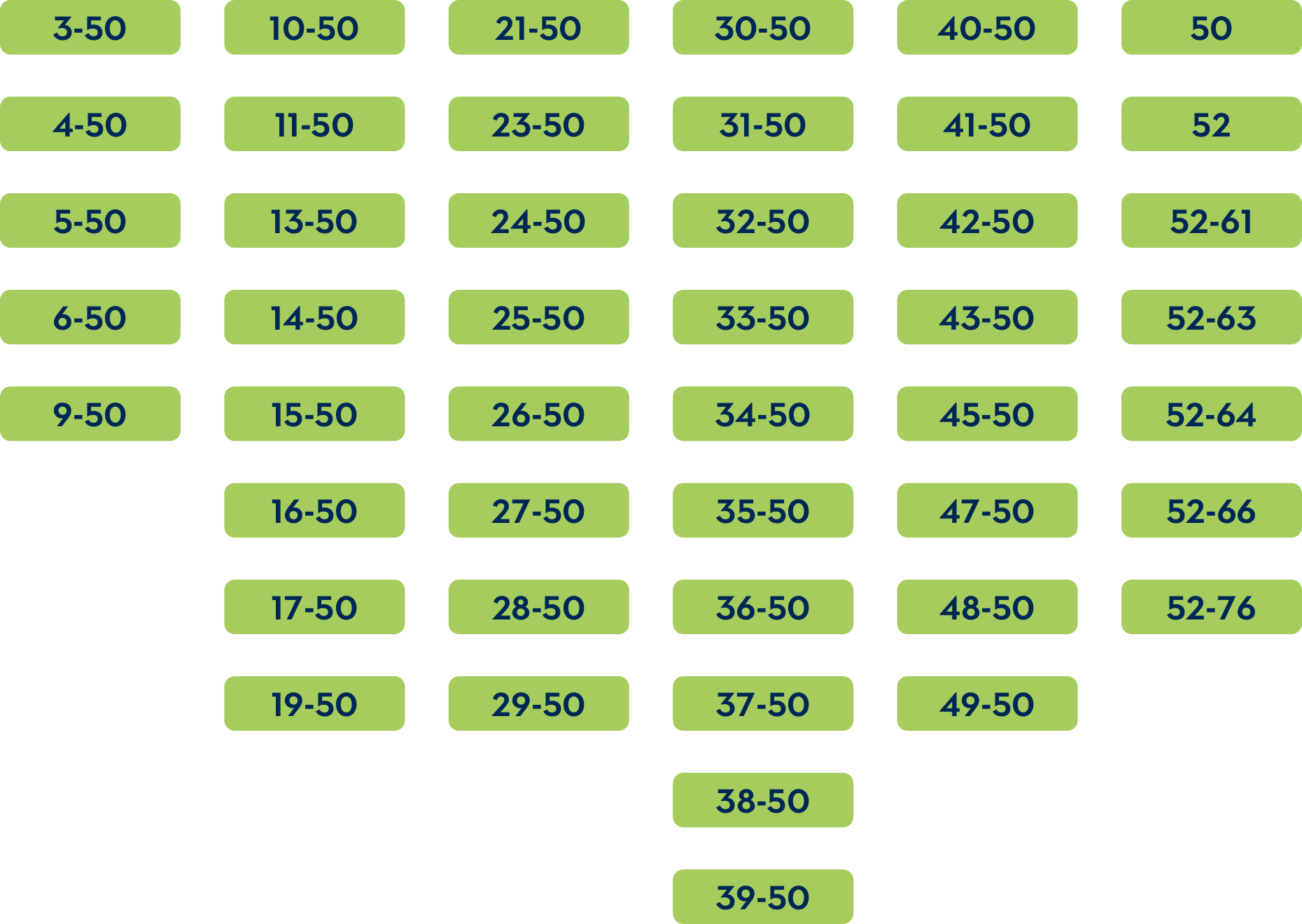
If a deletion of exons 52-58 or exon 52-77 occurs, skipping exon 51 is predicted to create a stop codon.
Patients with DMD who have a deletion of exon 52 may be treated with a therapy that skips either exon 51 or exon 53.
IMPORTANT SAFETY INFORMATION
EXONDYS 51 (eteplirsen) is indicated for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 51 skipping. This indication is approved under accelerated approval based on an increase in dystrophin in skeletal muscle observed in some patients treated with EXONDYS 51. Continued approval for this indication may be contingent upon verification of a clinical benefit in confirmatory trials.
IMPORTANT SAFETY INFORMATION
Hypersensitivity:
Hypersensitivity reactions, including bronchospasm, chest pain, cough, tachycardia and urticaria, have occurred in patients who were treated with EXONDYS 51. If a hypersensitivity reaction occurs, institute appropriate medical treatment and consider slowing the infusion or interrupting the EXONDYS 51 therapy.
Adverse Reactions:
Adverse reactions in DMD patients (N=8) treated with EXONDYS 51 30 or 50 mg/kg/week by intravenous (IV) infusion with an incidence of at least 25% more than placebo (N=4) (Study 1, 24 weeks) were (EXONDYS 51, placebo): balance disorder (38%, 0%), vomiting (38%, 0%) and contact dermatitis (25%, 0%). The most common adverse reactions were balance disorder and vomiting. Because of the small numbers of patients, these represent crude frequencies that may not reflect the frequencies observed in practice. The 50 mg/kg once weekly dosing regimen of EXONDYS 51 is not recommended.
The most common adverse reactions from observational clinical studies (N=163) seen in greater than 10% of patients were headache, cough, rash, and vomiting.
Before administration, please see the full Prescribing Information for EXONDYS 51 (eteplirsen).
INDICATION
EXONDYS 51 (eteplirsen) is indicated for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 51 skipping. This indication is approved under accelerated approval based on an increase in dystrophin in skeletal muscle observed in some patients treated with EXONDYS 51. Continued approval for this indication may be contingent upon verification of a clinical benefit in confirmatory trials.
References: 1. EXONDYS 51 [package insert]. Cambridge, MA: Sarepta Therapeutics, Inc. 2022. 2. Aartsma-Rus A, Van Deutekom JCT, Fokkema IF, et al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34(2):135-144. 3. Wood MJA, Gait MJ, Yin H. RNA-targeted splice-correction therapy for neuromuscular disease. Brain. 2010;133(pt 4):957-972. 4. Data on file. Sarepta Therapeutics, Inc. 5. Ciafaloni E, Fox DJ, Pandya S, et al. Delayed diagnosis in Duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. 2009;155(3):380-385. 6. Alfano LN, Charleston JS, Connolly AM, et al. Long-term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine (Baltimore). 2019;98(26):e15858. 7. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251-267. 8. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018;17(4):347-361. 9. Mercuri E, Signorovitch JE, Swallow E, et al; DMD Italian Group; Trajectory Analysis Project (cTAP). Categorizing natural history trajectories of ambulatory function measured by the 6-minute walk distance in patients with Duchenne muscular dystrophy. Neuromuscul Disord. 2016;26(9):576-583. Erratum in: Neuromuscul Disord. 2017;27(5):e1. 10. McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48(3):343-356. 11. Muntoni F, Domingos J, Manzur AY, et al; UK NorthStar Network. Categorising trajectories and individual item changes of the North Star Ambulatory Assessment in patients with Duchenne muscular dystrophy. PLoS One. 2019;14(9):e0221097. 12. Ricotti V, Ridout DA, Pane M, et al; UK NorthStar Clinical Network. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials. J Neurol Neurosurg Psychiatry. 2016;87(2):149-155. 13. McDonald CM, Henricson EK, Abresch RT, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018;391(10119):451-461. 14. Pane M, Coratti G, Brogna C, et al. Longitudinal analysis of PUL 2.0 domains in ambulant and non-ambulant Duchenne muscular dystrophy patients: how do they change in relation to functional ability? J Neuromuscul Dis. 2023;10(4):567-574. 15. McDonald CM, Gordish-Dressman H, Henricson EK, et al; CINRG investigators for PubMed. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: long-term natural history with and without glucocorticoids. Neuromuscul Disord. 2018;28(11):897-909. 16. Power A, Poonja S, Disler D, et al. Echocardiographic image quality deteriorates with age in children and young adults with Duchenne muscular dystrophy. Front Cardiovasc Med. 2017;4:82.
This information is intended for U.S. healthcare professionals only.